












コラム
SENDとFDA、そして産業界の動き
SENDの始まり
現在は多くの皆さんにその存在が認識されているSEND注1ですが、私が初めてSENDと出会ったのは、かれこれ15年ほど前のことです。当時はスイスに本拠を置く企業に勤務しており、その研究施設で使われていたコンピュータ・システムの所見入力画面にSENDという見慣れないデータ転送用のタブがついていたのを覚えています。勿論、当時SENDは米国でも義務化されていませんでしたが、すでにFDAはSENDの検討を開始しており、欧米のITベンダーは実験的な試みを始めていました。現在は一部の例外を除いてFDAに提出されるデータはすべて標準化された電子フォーマットでの提出が義務付けられています。
製薬メーカーにとっては、多くの場合、国内外の複数のCROを使っており、彼らのデータをSENDデータに作り変えなければならないため、膨大な作業が発生します。また、SENDデータをCROから受け取ることができたとしても、その品質の均一化、QCなど、データとしての保証は申請者である製薬メーカーが負うことになります。
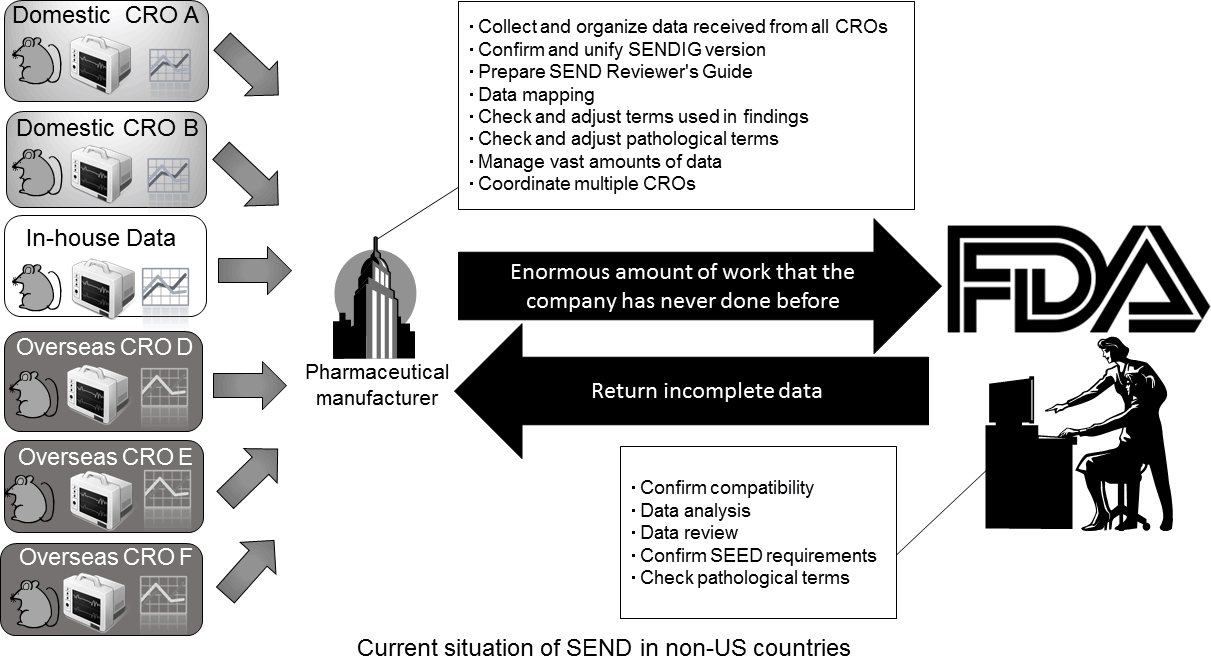
Reproduced with permission of the Japanese Society of Toxicologic Pathology from Anzai et al. "Establishment of the Global SEND Alliance (G-SEND) in Japan and efficient creation of electronic SEND datasets between CROs.", J Toxicol Pathol., 28(2): 57-64, 2015
ではなぜFDAがここまで多くの時間と予算をかけ、製薬メーカーに負担をかけてまで電子データ申請にこだわってきたか、その答えは彼らのIT5カ年計画、10カ年計画を見れば大義名分は理解できます。しかし、本音はもっと奥深いところにあると個人的には考えています。
大義名分と創薬に関わる壮大な思惑?
まず、大義名分ですが、新薬の申請データの評価には膨大な時間がかかるため、この時間の短縮は申請者・医療関係者・患者にとってベネフィットがあるということです。事実、私がFDAの審査官に聞いた話では、特に先行品との比較には多くの時間を費やしていたが、過去、現在のデータが統一電子フォーマットであれば容易にデータの比較検定が可能となり、審査は格段に早くなると語られていました。また、将来は市場にある医薬品の有害事象が発生した際に、迅速にデータ検索・解析できることもベネフィットの一つです。当然、類薬との比較解析も容易です。
さて、この大義名分とは別に、本音というべきか語られない目的があると個人的には見立てています。それはAIです。ちょうどSENDの検討が開始された当時は第二次AIブームから第三次AIブームへの過渡期にあたり、ビッグデータ、即ちマシーン・ラーニング(機械学習研究)が始まった頃です。その後、その一部である自動運転などは私たちの社会を大きく変えようとしています。当然、我々の分野においてもデータ・マイニングに代表されるバイオ・インフォーマティクスも発展しています。今後、医薬品開発においてもAIは欠くことのできないツールとなるでしょう。でも、そこには大きな課題があります。つまり、医薬品開発あるいは医薬品安全性・有効性評価に過去の膨大なデータをAIが使うとすれば、それは機械検索可能な標準化されたデータでなければならないということです。それがSENDです。
FDAが開始したのは非臨床試験ばかりでなく、当然ながら臨床試験データの電子標準化も同時に進めています。おそらく10年後、20年後には医薬品開発を激変させるスーパー・バリュー・ビックデータとなるでしょう。AIの発展もめざましいものがあります。AIがビッグデータの情報をもとに、全く新しい有効性と安全性の高い薬をデザインする日も遠くないでしょう。この予測に立てば米国の本音は新薬開発国家としての地位を維持するための知的財産戦略と見ることができます。つまり、SENDは単なる電子データ化についての規制ではなく、将来は創薬・安全性研究にも大きく関わってくる可能性を持っていることを認識しておく必要があると思います。
製薬企業のSEND対応はどうなっている?
さて、日本に限った話ではありませんが、製薬企業、CROによる現実的なSEND対応については、一部の先行する製薬企業とCROを除けば、SENDの学習段階がやっと終わったといったところでしょう。学習機関としてはCDISC注2やPhUSE注3といった団体があります。製薬企業の多くはこれらの組織に属して勉強を続けていますが、現実的なSEND対応においては明暗を分ける形となっていると思います。それはその企業がSENDをIT課題と捉え莫大な設備投資をして対応したか、申請技術課題と捉え開発品毎のRSP注4を使った個別予算アウトソーシング課題と捉えたかによる結果の違いからも見えてきます。
答えは明らかで、多くの製薬企業は申請用試験の60%以上、場合によっては80%以上をCROに委託しており、製薬企業内部で行われるGLP試験はそれほど大きくありません。これにも関わらず、数少ない内製試験のために数億円規模の設備投資と人的資源を投入し、自らSENDデータセットを作成する経営的メリットはありません。また、どんなにお金をかけて高額な設備投資をしたところで、結局は非臨床試験の申請資料を作るのは安全性部門であり、データマネージメント部門ではありません。
一方、莫大な設備投資と人的投資を避け、開発単位でのSEND対応を行う方法は現在主流となっています。この場合、SENDデータセットは開発毎の予算(概ね試験費用の10%から20%をSEND予算として追加)でまかないます。当然、開発プロジェクト繁忙期、閑散期に関係なくかかってしまう固定費(設備投資・人的投資)は発生しませんので、経済性が高く、更に専門家によるサポートも受けられます。問題点としては実績のあるRSPやCROが少ないということですが、この手法をとっている製薬企業の多くはこれらRSPあるいはRSPの支援を受けたCROと包括的な契約を早い段階で締結しているようです。これらの製薬企業はいくつもの開発品を抱えており、SENDに設備投資や人的投資をしている時間的余裕がなく、いかに早く確実なSENDデータセットを労せずに作り上げるか、つまり、本業に忙しい製薬メーカーほどこの傾向が強いように思います。幸い、その方が成功しているようです。ぶっちゃけSENDは製薬企業にとって本業ではありません。
CROの新たな動きと製薬メーカーにとっての希望の光
一方CROの動きとしては、製薬企業からの要望に対応できるよう、SENDデータ作成基地となるCROを定め、データ変換を共通のツールとプロセスで実施するコンソーシアム的な非営利団体であるG-SEND注5が発足しています。ほとんどのCROは受託した試験(契約試験)のSENDデータ作成しか行いませんが、この集団では中心となるCROがSEND基地として製薬メーカーの内製試験のSENDデータ作成も行なっています。また、RSPのサポートを集団で受けることが可能で、最新のFDA情報、技術情報の共有化、G-SENDによるSENDデータ作成プロセスの標準化、品質向上など、SENDデータを受け取る製薬メーカーにとっても、CRO間でバラツキのない、均一かつ高品質のSENDデータセットを受け取れるなど多くの利点があります。CDISCやPhUSEが我々のSEND学習機関であるのに対し、G-SENDはSEND実践機関と言えるでしょう。
現在、G-SENDは日本のCROを中心に16社、韓国、台湾、シンガポール、米国、スイスの6カ国のメンバーで構成されています。また、同業者が多いため、厳しいコンソーシアム管理が行われ、RSPによるフル・サポートも受けています。勿論、単独でSEND対応を行なっているCROも存在しますが、規制対応ビジネスモデルとしては集団対応が優れていることは間違いないでしょう。また、PhUSEとFDAによる研究においても、複数のCROが協働するマルチサイト・スタディーなどではSENDメインCROを定めてSEND対応するシナリオも発表されています。いずれにしてもG-SENDというビジネスモデルでCROのSEND対応が組織化されたことは大きな意味を持っていると思います。また、このことは製薬メーカーにとっては朗報(希望の光?)であることは間違いないでしょう。
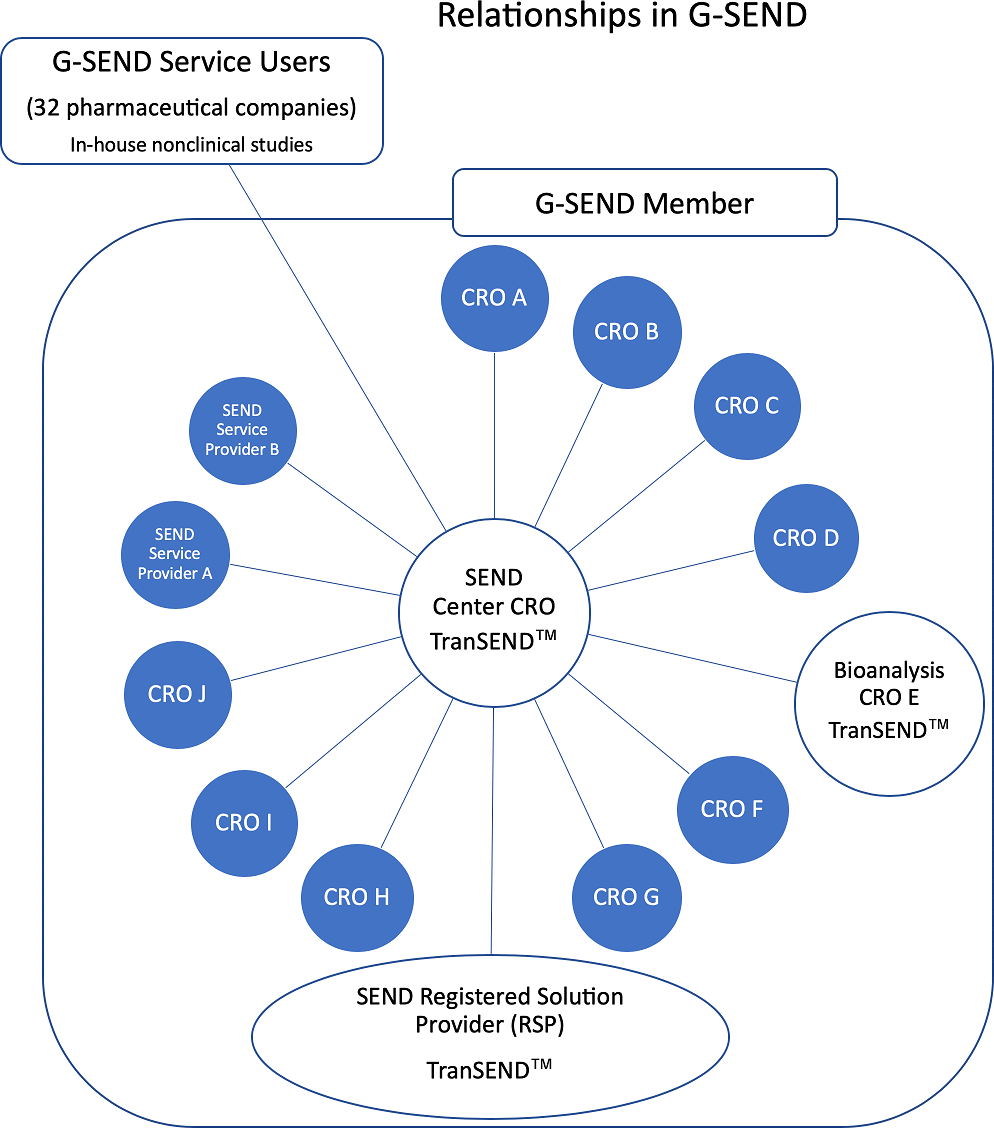 Reproduced with permission of the Japanese Society of Toxicologic Pathology from Anzai et al. "Establishment of the Global SEND Alliance (G-SEND) in Japan and efficient creation of electronic SEND datasets between CROs.", J Toxicol Pathol., 32(2): 119-126, 2019.
Reproduced with permission of the Japanese Society of Toxicologic Pathology from Anzai et al. "Establishment of the Global SEND Alliance (G-SEND) in Japan and efficient creation of electronic SEND datasets between CROs.", J Toxicol Pathol., 32(2): 119-126, 2019.
製薬企業は今まで、CROへ試験を委託する際に、技術力、実績、コストに重きを置いてきましたが、現在はFDAの審査を確実に通過できる高品質なSENDデータを提供できるCRO(あるいはコンソーシアム・メンバー)であるか否かが新たな選択基準として加わっています。
今後は日本、ヨーロッパ、アジアの電子データ申請の動きも注目し、いつの日か再び皆さんに情報提供させていただく機会があることを期待しています。
令和元年6月
昭和大学医学部
安齋享征
注1: Standard for Exchange of Nonclinical Data
注2: CDISC: Clinical Data Interchange Standards Consortium
注3: PhUSE: Pharmaceutical Users Software Exchange
注4: CDISCに登録されているSEND Registered Solution Provider
注5: G-SEND; Global SEND Alliance
参考論文
- Establishment of the Global SEND Alliance (G-SEND) in Japan and efficient creation of electronic SEND datasets between CROs. https://www.ncbi.nlm.nih.gov/pubmed/31092979
- Responses to the Standard for Exchange of Nonclinical Data (SEND) in non-US countries. https://www.ncbi.nlm.nih.gov/pubmed/26028814
- Specific pathologist responses for Standard for Exchange of Nonclinical Data (SEND). https://www.ncbi.nlm.nih.gov/pubmed/28798527
![]()